généralité
Craniosynostose est le terme par lequel les médecins indiquent une anomalie du crâne due à la fusion prématurée d'une ou plusieurs sutures crâniennes.
Les sutures crâniennes sont les articulations fibreuses qui unissent les os de la voûte crânienne (c.-à-d. Les os frontal, temporal, pariétal et occipital).
La craniosynostose peut être un phénomène isolé (craniosynostose non syndromique) ou le résultat de certaines conditions morbides particulières (craniosynostose syndromique). Parmi les affections morbides à l'origine de la fusion prématurée des sutures crâniennes, les plus connues sont: le syndrome de Crouzon et le syndrome d'Apert.
Avec la fusion prématurée des sutures crâniennes, les structures encéphaliques ne disposent pas d’un espace suffisant pour leur croissance. Cela a diverses conséquences, notamment l'augmentation de la pression intracrânienne (hypertension intracrânienne).
Un diagnostic rapide et précis permet de planifier un traitement ad hoc . Ce dernier est de type chirurgical et a pour objectif final la séparation précoce des sutures soudées.
Rappels d'anatomie du crâne humain
Doté d'os et de cartilages, le crâne est la structure squelettique de la tête qui constitue le visage et protège le cerveau, le cervelet, le tronc cérébral et les organes sensoriels.

Pour simplifier l'étude et la compréhension du crâne, les anatomistes ont pensé à le scinder en deux compartiments, appelés neurocrâne et splancnocrâne .
neurocranium
Le neurocrâne est la région du crâne supérieur contenant le cerveau et certains des principaux organes sensoriels. Ses os les plus importants - strictement plats - sont les os frontal, temporal, pariétal et occipital; ceux-ci, ensemble, forment ce qu'on appelle la voûte crânienne .
splanchnocrâne
Le splanchnocranium, ou facial facial massif, est la région antéro-inférieure du crâne, composée d'os uniformes et inégaux. Il représente la structure squelettique du visage, il contient donc des éléments osseux tels que la mâchoire, la mâchoire supérieure, les pommettes, l'os nasal, etc.
Qu'est-ce que la craniosynostose?
La craniosynostose est une anomalie rare du crâne caractérisée par une forme de tête peu naturelle due à la fusion prématurée d'une ou plusieurs sutures crâniennes . Les sutures crâniennes sont les articulations fibreuses qui unissent les os de la voûte crânienne (c.-à-d. Les os frontal, temporal, pariétal et occipital).

Depuis le site: //www.wkomsi.com/
QUAND LA FERMETURE DES SUTURES CRANIENNES DEVRAIT-ELLE AVOIR?
Dans des conditions normales, la fusion des sutures crâniennes a lieu au cours de la période postnatale (NB: certains processus se terminent même à 20 ans). Ce long processus de fusion permet au cerveau de se développer et de se développer correctement.
Si, comme dans le cas de la craniosynostose, la fusion a lieu trop tôt - donc durant la période prénatale, périnatale * ou précoce, les éléments encéphaliques (cerveau, cervelet et tronc cérébral) et certains organes des sens (yeux notamment) une altération de la forme et de la croissance.
Causes
Le processus physiopathologique qui détermine la craniosynostose est la fusion prématurée des sutures crâniennes .
Ce processus peut représenter un phénomène isolé - où "isolé" signifie qu'il n'est associé à aucun état morbide particulier - ou peut être la conséquence de certains syndromes particuliers, presque toujours de nature génétique.
À la lumière de cela, les médecins ont pensé classer la craniosynostose en deux catégories:
- Craniosynostose non syndromique . Le terme non syndromique indique que l'anomalie crânienne n'est associée à aucune pathologie ou autre défaut physique.
- La craniosynostose syndromique . Le terme syndromique signifie que la malformation crânienne est le résultat d'un syndrome particulier, dans la plupart des cas de type génétique.
CRANIOSINOSTOSE NON SYNDROMIQUE
Les médecins et les chercheurs n'ont pas encore établi les causes de la craniosynostose non syndromique.
Ils ont proposé diverses hypothèses - y compris l’influence de facteurs environnementaux ou de problèmes analogues aux hormones - mais aucune de ces théories n’a été confirmée par les résultats expérimentaux.
Par conséquent, pour comprendre l'origine exacte de l'anomalie, des études supplémentaires sont nécessaires.
CRANIOSINOSTOSE SINDROMICA
Selon les dernières recherches médicales, il existe plus de 150 syndromes différents, tous très rares, susceptibles de provoquer une craniosynostose.
Parmi ces syndromes, les plus connus et les plus communs sont:
- Syndrome de Crouzon . Résultat de mutations spécifiques dans les gènes FGFR2 (chromosome 10) et FGFR3 (chromosome 4), cet état morbide affecte un nouveau-né tous les 60 000 habitants et implique la présence d'anomalies exclusivement au niveau de la tête et du visage.
- Syndrome d'Apert . Elle est principalement due à des mutations du gène FGFR2 (identique au syndrome de Crouzon) et affecte un nouveau-né toutes les 100 000 personnes ou plus.
Contrairement au syndrome de Crouzon, les altérations génétiques de FGFR2 sont telles que les malformations touchent non seulement le crâne et le visage, mais également les mains et les pieds.
- Syndrome de Pfeiffer . Il résulte de mutations dans le gène "habituel" de FGFR2 et d'un gène ayant des fonctions similaires, appelé FGFR1 (chromosome 8). La particularité de ces mutations est que, outre les déformations du crâne et du visage, elles déterminent également: la syndactylie, la brachydactylie, les pouces et les gros orteils (disproportionnés par rapport aux autres doigts).
Le syndrome de Pfeiffer a une incidence d'un nouveau-né sur 100 000.
- Syndrome de Saéthre-Chotzen . Il s’agit d’une maladie génétique touchant un nouveau-né tous les 50 000 habitants environ. Provoque diverses malformations au niveau du crâne, du visage, des mains et des pieds. Certaines mutations spécifiques du gène TWIST1, situées sur le chromosome 7, sont responsables de l’apparition du syndrome de Saethre-Chotzen.
ÉPIDÉMIOLOGIE DE LA CRANIOSINOSTOSE
Selon les statistiques les plus récentes, il semblerait qu'un enfant d'environ 1800-3000 soit atteint de craniosynostose.
En ce qui concerne le sexe le plus affecté, plusieurs études cliniques ont montré que 3 patients sur 4 étaient des hommes. La raison pour laquelle la craniosynostose est plus répandue dans la population masculine est totalement inconnue.
Facteurs de risque de craniosynostose.
- Faible poids à la naissance
- Naissance prématurée
- Âge paternel avancé
- Tabagisme maternel pendant la grossesse
Symptômes et Complications
La plupart des symptômes pouvant être observés en présence de craniosynostose sont dus à une augmentation de la pression dans le crâne . En médecine, l'augmentation de la pression dans le crâne est appelée hypertension intracrânienne ou hypertension intracrânienne .
En présence de craniosynostose, l'hypertension intracrânienne est une conséquence du fait que le cerveau et d'autres structures situées à l'intérieur du crâne ne disposent pas de l'espace adéquat pour se développer. Ils vont donc pousser sur les structures osseuses de la tête.
Cela dit, il est important de rappeler que, si les sutures crâniennes impliquées sont nombreuses ou si la maladie n'est pas traitée à temps, la craniosynostose peut entraîner un développement réduit des capacités cognitives et un faible QI.
SYMPTÔMES DE L’HYPERTENSION ENDOCRANIQUE
Les symptômes possibles de l'hypertension intracrânienne sont:
- Céphalée persistante La situation empire généralement le matin et le soir.
- Problèmes de vision. Ils consistent en une vision double, une vision floue et une vision floue.
- vomissement
- irritabilité
- Les yeux gonflés ou proéminents
- Difficulté à suivre le mouvement des objets
- Problèmes d'audition
- Problèmes respiratoires
- Altérations de l'état mental
- papilledema
Le nombre de sutures crâniennes impliquées dans le développement de la craniosynostose a une influence significative sur la présence d'hypertension intracrânienne.
Par exemple, les médecins ont observé que l'implication d'une seule suture crânienne induit une hypertension intracrânienne chez 15% des patients; tandis que l'implication d'au moins deux points de suture entraîne une augmentation de la pression dans le crâne chez au moins 60% des patients.
En présence d'une forme légère de craniosynostose, l'hypertension endocrânienne commence à poser problème, provoquant la symptomatologie susmentionnée, d'environ 4 à 8 ans de la vie.
SIGNES DE CRANIOSINOSTOSE
Parmi les signes de craniosynostose, les plus courants sont:- Formations de crêtes rigides le long des sutures crâniennes
- Anomalies au niveau de la fontanelle crânienne
- Tête dont les dimensions ne sont pas proportionnées au reste du corps
TYPES DE CRANIOSINOSTOSE
La forme de la tête des patients atteints de craniosynostose dépend des sutures crâniennes qui se sont refermées prématurément.
Les médecins ont donc constaté qu'il convenait de distinguer les craniosynostoses selon divers types de sutures crâniennes.
Les types de craniosynostose sont:
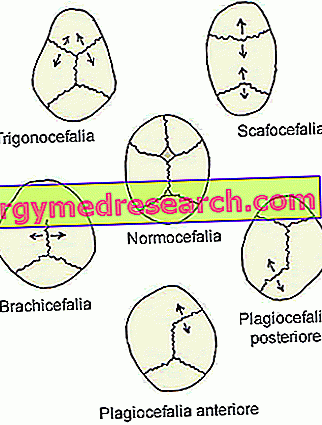
- Synostose sagittale ( dolichocéphalie ou scafocéphalie ). C'est le type le plus commun de craniosynostose; en fait, il caractérise environ la moitié des cas cliniques.
Sa présence coïncide avec la fermeture prématurée des sutures crâniennes sagittales situées dans la partie supérieure du crâne, entre les os pariétaux.
De //en.wikipedia.org/wiki/Plagiocephaly
- Craniosynostose coronale ( brachycéphalie ) Il s'agit du deuxième type de craniosynostose le plus répandu. il présente environ un cas clinique sur quatre.
Son apparition implique la fusion prématurée des sutures coronales, qui s'étendent entre l'os frontal et les os pariétaux.
- Synostose métopique ( trigonocéphalie ). Il s'agit d'un type de craniosynostose plutôt rare, qui ne distingue que 4 à 10% des cas.
Son apparition coïncide avec la fusion prématurée de la suture métopique (ou frontale), qui va du nez au sommet de la tête, séparant l'os frontal en deux. En règle générale, cette suture s'ossifie naturellement au cours de la sixième année de vie.
- Sinostose Lambdoïde ( plagiocéphalie ). C'est le type le plus rare de craniosynostose. En fait, il ne distingue que 2 à 4% des cas cliniques.
Sa présence implique la fusion précoce de la suture lambdoïde, située entre les os pariétaux et l'os occipital, à l'arrière de la tête.

COMPLICATIONS
En plus d'affecter le développement intellectuel, une craniosynostose non traitée peut déterminer:
- Le soi-disant syndrome d'apnées obstructives du sommeil .
- Altérations faciales permanentes au niveau des yeux et des oreilles en particulier.
- Déformations permanentes à la base du crâne (par exemple, la malformation ou le syndrome d'Arnold-Chiari).
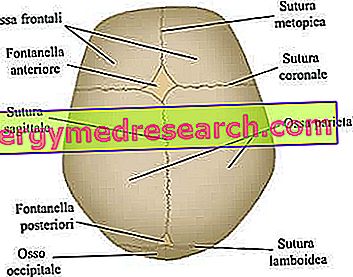
Les principales sutures crâniennes impliquées dans le processus de craniosynostose. Depuis le site web: www.sciencebasedmedicine.org
- L'hydrocéphalie .

diagnostic
Pour diagnostiquer la craniosynostose, un examen objectif, une évaluation des antécédents cliniques et des images radiologiques fournies par les rayons X ou le scanner à la tête sont essentiels.
Si le craniosynostose était de type syndromique, il est également important d'établir la condition morbide qui a déterminé son apparition. Par conséquent, les médecins pourraient avoir recours à des tests sanguins et, surtout, à un conseil génétique .
EXAMEN OBJECTIF
L'examen objectif consiste en une analyse minutieuse par le médecin des signes cliniques présents sur la tête du sujet, suspectés de souffrir de craniosynostose.
Généralement, un pédiatre est impliqué dans cette vérification diagnostique importante.
HISTORIQUE CLINIQUE
L'évaluation des antécédents cliniques est importante à des fins de diagnostic car elle inclut des questions relatives aux facteurs de risque de craniosynostose.
Ainsi, le médecin (généralement toujours un pédiatre) examinera si:
- Le bébé est né prématuré ou de poids faible.
- Quel était l'âge du père au moment de la conception?
- Si la mère a fumé pendant la grossesse.
EXAMENS RADIOLOGIQUES
La radiographie et le scanner à la tête servent avant tout à confirmer le diagnostic et à montrer au médecin quelles sutures crâniennes ont soudé de manière prématurée.
La connaissance des sutures crâniennes impliquées permet de planifier le traitement chirurgical le plus approprié.
traitement
La craniosynostose ne peut être guérie que par une intervention chirurgicale .
Ce dernier consiste en une opération de séparation des sutures de fusel crâniennes précocement entre elles.
L’objectif thérapeutique final de la chirurgie est de fournir aux structures cérébrales et à certains organes sensoriels, tels que les yeux, cet espace nécessaire au développement et au fonctionnement optimal.
MEILLEUR MOMENT À INTERVENIR
Il n’ya pas d’accord total entre les médecins sur le meilleur moment pour pratiquer une chirurgie de craniosynostose.
Selon certains experts, la période idéale pour l'opération serait la fin de l'enfance, lorsque le risque de rechute (c'est-à-dire une deuxième fusion prématurée de sutures crâniennes) est moindre. En cas de récidive, en effet, l'intervention doit être répétée et ceci n'est pas recommandé compte tenu de la finesse de la procédure.
Selon d’autres experts, le moment le plus approprié serait celui de la petite enfance (entre 6 et 12 mois), lorsque le crâne n’est pas encore complètement ossifié et que les os servent encore de modèle. La possibilité de modeler les os (malléabilité) permet de résoudre toute anomalie morphologique des os eux-mêmes, ce qui pourrait provoquer de graves défauts esthétiques et des problèmes fonctionnels (de la mâchoire ou des yeux) à un âge plus avancé.
APPROCHES DE CHIRURGIE POSSIBLES
Il existe deux approches chirurgicales différentes: la chirurgie traditionnelle, également appelée «à l'air libre» et la chirurgie endoscopique.
- Chirurgie traditionnelle (ou "en plein air") .
Cela implique une anesthésie générale (le patient est donc inconscient pendant toute la procédure) et la pratique d'une incision chirurgicale à la tête, exactement là où les images radiologiques ont montré l'anomalie crânienne.
À travers l'incision sur la tête, le chirurgien opérant (un neurochirurgien) retire l'os anormal et le confie à un spécialiste en chirurgie cranofaciale, qui le modifie et lui donne une forme permettant le développement normal des structures encéphaliques.
Après modification, le neurochirurgien remplace l'os dans sa position d'origine et ferme l'incision avec des sutures.
Comme beaucoup de chirurgies traditionnelles, l'opération "à ciel ouvert" est plutôt invasive; Cependant, il est avantageux de pouvoir modifier la structure de l'os de manière précise et avec de bons résultats.
- Chirurgie endoscopique .
Cela implique l'utilisation d'un endoscope, un outil similaire à un tube flexible, équipé d'une caméra à fibre optique à une extrémité et connecté à un moniteur.
D'un point de vue opératoire, il consiste à insérer l'endoscope dans une ouverture pratiquée sur le crâne et à séparer, à l'aide de l'endoscope lui-même, la suture fusionnée (suture).
Le neurochirurgien parvient à s’orienter à l’intérieur de la tête, grâce aux images que la caméra projette sur le moniteur connecté en externe.
La chirurgie endoscopique est décidément moins invasive que l’opération "à champ ouvert" (même la période d’hospitalisation la plus courte), mais elle présente deux inconvénients: elle n’est indiquée que pour les patients de quelques mois (6 en général), qui possèdent des os qui peuvent encore être modelés; il est plus à risque de rechute.
PHASE POSTOPÉRATOIRE
En règle générale, un patient souffrant de craniosynostose, qui a subi une intervention chirurgicale, doit rester à l'hôpital pendant environ 4-5 jours après l'opération. Pendant ce temps, le neurochirurgien et les membres de son personnel contrôlent périodiquement les paramètres vitaux et vérifient que tout se passe bien.
Après la démission, une série de contrôles périodiques est prévue, qui sont d’abord semestriels puis, avec la croissance du patient, tous les ans.
pronostic
Le pronostic dépend de divers facteurs, notamment:
- Causes qui ont provoqué la craniosynostose. Certaines maladies génétiques responsables de cette anomalie sont très graves et de mauvais pronostic.
- La position des sutures du fusel prématurément. Si les sutures se trouvent dans des positions qu'il est "peu pratique" d'atteindre pour le neurochirurgien, l'intervention du craniosynostosis devient compliquée et peut ne pas donner les résultats souhaités.


